|
Imitating Rube
Does the human clotting system lend itself
to the same kind of analysis? At its core, the actual mechanism
of clotting is remarkably simple. A fibrous, soluble protein
called fibrinogen ("clot-maker") comprises about 3%
of the protein in blood plasma. Fibrinogen has a sticky portion
near the center of the molecule, but the sticky region is covered
by little amino acid chains with negative charges. Because like
charges repel, these chains keep fibrinogen molecules apart.
When a clot forms, a protease (protein-cutting)
enzyme clips off the charged chains. This exposes the sticky
parts of the molecule, and suddenly, fibrinogens (which are now
called fibrins) start to stick together, beginning the formation
of a clot.
The protease that cuts off the charged
chains is called thrombin. So, just like the lobster clotting
system, the heart of the reaction involves just two molecules:
fibrinogen and thrombin. But, unlike the lobster, there's a lot
more to this machine. It turns out that thrombin itself exists
in an inactive form called prothrombin. So it, just like fibrinogen,
has to be activated before it can start the clotting process.
What activates prothrombin? Here's where life gets really interesting.
Prothrombin, a protease itself, is activated by another
protease called Factor X which clips of part of the inactive
protein to produce active, clot-forming thrombin. OK, so what
activates Factor X? Believe it or not, there are still more proteases,
two of them, actually, called Factor VII and Factor IX, that
can switch on Factor X. What switches them on? Here's where a
good teacher goes the the blackboard, and so will I:
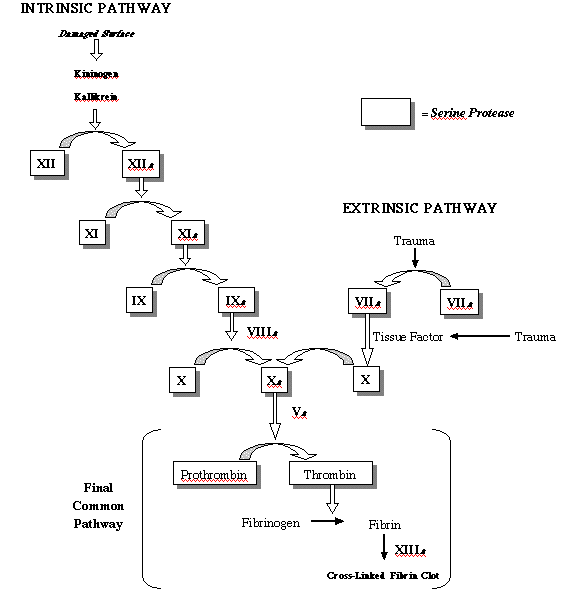
Now, "beauty" is a word that
most people wouldn't think to put in the same sentence with "biochemistry,"
but the biochemistry of this pathway is beautiful indeed. Start
at the bottom, with the conversion of soluble fibrinogen into
clot-forming fibrin. As you look up, you can trace this process
to one of two different external stimuli, both of which make
good sense. At the upper left, the pathway can be started with
damage to a cell surface, something that happens whenever blood
is exposed to the air or a foreign object at the surface of a
wound. At the upper right, tissue factor, a soluble protein found
in most tissues but not in the bloodstream, activates the pathway.
This is where clotting starts from an internal hemorrage, a broken
vessel within the tissues of the body. So, both of these ultimate
stimuli lead to the same set of clot-forming proteins (Factor
X, thrombin, and fibrin), but neither does it directly. Instead,
each activates a "cascade" of intermediate factors,
nearly all of them proteases, which eventually activate clot
formation.
It sure does look pretty, but why a cascade?
Why couldn't we have a simpler pathway, like the lobster, where
something like tissue factor activated clotting directly? Well,
we could, but a complex pathway, even if it drives biochemistry
students to distraction, has advantages of its own. For one thing,
the multiple steps of the cascade amplify the signal from
that first stimulus. If a single active molecule of Factor XII
could activate, say, 20 or 30 molecules of Factor XI, then each
level of the cascade would multiply the effects of a starting
signal. Put 5 or 6 steps in the cascade, and you've amplified
a biochemical signal more than a million times. Clotting with
fewer steps would still work, but it would take longer to produce
a substantial clot, and would be much less responsive to smaller
injuries.
Michael Behe is in awe of the the intricate
complexity of this system, and so am I. And he is also correct
in pointing out that if we take away part of this system, we're
in trouble. Hemophiliacs, for example, are unable to synthesize
the active form of Factor VIII. This means that they are unable
to complete the final step of one of the pathways, and that's
why hemophilia is sometimes known as the bleeder's disease. Defects
or deficiencies in any of the other factors are equally serious.
No doubt about it - clotting is an essential function and it's
not something to be messed with. But does this also mean that
it could not have evolved? Not at all. The key to understanding
the evolution of this intricate system, as Russell Doolittle
has pointed out, is the fact that the clotting factors share
an exquisite and revealing similarity.
Building the Machine
To paraphrase Darwin, the notion that
evolution could have produced a system as intricate as the blood
clotting cascade seems, we might freely confess, "absurd
in the highest possible degree." This is especially true
if you believe, as Behe seems to, that clotting is not possible
until the entire cascade of factors is assembled.
But we already know that evolution doesn't
start from scratch, and it doesn't need fully-assembled systems
to work. Remember the lobster system as an example. Blood clotting
evolved there from two pre-existing proteins, normally found
in separate compartments of the body, that had a fortuituous
interaction when damage to a blood vessel brought them together.
Once that interaction was established, natural selection did
the rest.
Could something like this have happened
here?
Remember, we're not starting from nothing.
We're starting about 600 million years ago in a small pre-vertebrate.
with a low-volume low-pressure circulatory system. Just like
any small inverterbate with a circulatory system, our ancestral
organism would have had a full compliment of sticky white cells
to help plug leaks. In addition, that ancestral system would
have had something else. Most of the time, hemorrage starts with
cell injury, meaning that cells are broken in the vicinity of
a wound and their contents are dumped out. That means, among
other things, that all of a cell's internal signalling molecules
are suddenly spilled out into the damaged vascular system. Included
among the contents are a whole slew of internal signalling molecules,
including prominent ones like cyclic adenosine monophosphate
(abbreviated: cAMP), all dumped into the tissue surrounding a
wound.
Why would a sudden gusher of cAMP in
a wound be significant? Well, it turns out that vertebrates use
cAMP as a signalling molecule to control the contractions of
smooth muscle cells, the very sort of muscle cells that surround
blood vessels. Therefore, the release of internal cAMP from broken
cells would automatically cause smooth muscles around a broken
vessel to contract, limiting blood flow and making it more likely
that the blood's own sticky white cells would be able to plug
the leak. That means that we already have some ability to limit
damage and plug leaks in a primitive, low-pressure system. Not
a bad place to begin.
Our next step is to consider the nature
of blood itself. For reasons relating to osmotic pressure, the
tendency of water to move across cell membranes, blood plasma
is a viscous, protein-laden solution. And it's also important
to note that the extracellular environment of ordinary tissue
is very different from blood. These spaces are laden with protein
signals, insoluble matrix molecules, and extracellular proteases
that cut and trim these molecules to their final shapes and sizes.
In fact, such proteases constitute one of the major forms of
extracellular signalling. So the tissues of our ancestral vertebrate
would be laden with protein-cutting enzymes for reasons completely
unrelated to clotting.
Keeping all of this in mind, what would
happen when a blood vessel broke in such an organism?
Well, protein-rich plasma flows into
an unfamiliar environment, and sticky white cells quickly "glom"
up against the fibers of the extracellular matrix. Tissue proteases,
quite accidentally, are now exposed to a new range of proteins,
and they cut many of them to pieces. The solubility of these
new fragments vary. Some are more soluble than the plasma proteins
from which they were trimmed, but many are much less soluble.
The result is that clumps of newly-insoluble protein fragments
begin to assumulate at the tissue-plasma interface, helping to
seal the break and forming a very primitive clot. (Could one
object that this is too primitive and too nonspecific to work?
That it wouldn't be sufficient to seal breaks? Well, it turns
out that you can't make this objection for the very simple reason
that this is pretty much the clotting mechanism used today by
a large number of invertebrates. Works for them, and therefore
there is no reason why it wouldn't have worked for the ancestors
of today vertebrates, either!)
Now we get down to business. A mutation
duplicates an existing gene for a serine protease, a digestive
enzyme produced in the pancreas. Gene duplications happen all
the time, and they are generally of such little importance that
they are known as "neutral" mutations, having no effect
on an organism's fittness. However, the original gene had a control
region that switched it on only in the pancreas. During the duplication,
the control region of the duplicate is damaged so that the new
gene is switched on in both the pancreas and the liver.
As a result, the inactive form of the enzyme, a zymogen, is relesased
into the bloodstream.
This causes no problem for the organism
- most pancreatic proteases are inactive until a small piece
near their active sites can be cut away by another protease.
However, when damage to a blood vessel allows plasma to seep
into tissue, suddenly our previously inactive plasma serine
protease is activated by tissue proteases, increasing the overall
protein-cutting activity at the site of the hemorrage. Blood
clotting is enhanced, so our duplicate gene (with the mistargeted
protein) is now favored by natural selection.
That plasma protease gene is now subject
to the same witches' brew of copying errors, rearrangements,
and genetic reshuffling that affect the genes for every other
cellular protein. Over time, bits and pieces of other genes are
accidentally spliced into the plasma protease sequence. Because
the selective value of the plasma protease is pretty low (it
doesn't help clotting all that much), most of these changes make
very little difference. But one day, through a well-understood
process called "exon shuffling," a DNA sequence known
as an "EGF domain" is spliced into one end of the protease
gene. EGF stands for epidermal growth factor, a small protein
used by cells throughout the body to signal other cells. EGF
is so common that just about every tissue cell has "receptors"
for it. These receptors are cell surface proteins shaped in such
a way that they bind EGF tightly.
The fortuitious combination of a EGF
sequence with the plasma protease changes everything.
In a flash, the tissue surrouding a broken
blood vessel is now teeming with receptors that bind to the new
EGF sequence on our serum protease. As a result, high concentrations
of the circulating protease bind directly to the surfaces of
cells near a wound. The proteases are activated in the same way,
but now their proteolytic activities are highly localized. The
production of a clot of insoluble protein fragments is now faster
and more specific than ever. Organisms with the new EGF-protease
can clot their blood much more quickly than before, and therefore
are favored by natural selection. To emphasize its role in the
clotting process, that cell surface protein with the EGF receptor
is called Tissue Factor.
What happens next? Well, remember the
case of the lobster in which a duplicate of a circulating protein
(vitellogenin) became specialized to produce a clot-forming protein
(lobster fibrinogen)? Once we have a situation in which every
hemorrage activates a protease bound to tissue receptors, a gene
duplicate of one of the major plasma proteins would then be under
strong selective pressure to increase its ability to interact
with the bound protease. Fibrinogen, the soluble protein that
now is now the primary target of proteolysis in the clotting
cascade, clearly arose in this way. Natural selection would favor
each and every mutation or rearrangement that increased the sensitivity
of fibrinogen to the plasma protease, dramatically enhancing
the ability of the new protease to form specific clots of insoluble
protein.
There is no doubt that these three steps,
each one supported by classic Darwinian mechanisms, would have
been sufficient to fashion a rudimentary clotting system. This
would leave us with system in which circulating plasma contains
both an inactive serine protease and its fibrinogen target. The
protease would activated by contact with tissue factor, and the
active protease, in turn, would cleave sensitive sites in fibrinogen
to form a clot. This system wouldn't be nearly as quick, as responsive,
or as sensitive as the current system of vertebrate clotting,
but it would work a little better than the system that preceeded
it, and that's all that evolution requires.
Adding Complexity
Could evolution take this rudimentary
system and produce a multilayered cascade of factors? Just watch.
Most serine proteases, including trypsin and thrombin, are auto-catalytic.
That means that some extent they can activate themselves, in
many cases by cleaving a few amino acids to switch on their active
sites. So, we could diagram the actual functions of our ancestral
plasma protease (which we'll call protease A) like this:
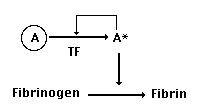
As we have seen, the inactive form of
the protease (A) is changed into the active form (A*) when two
things happen: it is bound to tissue factor (TF) and it is activated
by tissue proteases, including our protease itself (that's the
autocatalytic part). This means - and this is important - that
our protease is actually involved in cutting two things:
Fibrinogen, and also itself, converting A's inactive precursor
protein into A*.
Now, let's suppose that a gene duplication
occurs in the gene for our protease, producing a new (B) version
of the gene:
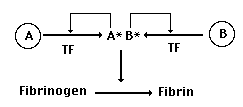
At first, just like most gene duplications,
this is no big deal. Proteins A and B are identical. Each can
bind to TF, each can cleave fibrinogen into fibrin, and each
can activate itself or its sister serum protease. So nothing
has really changed - we've just got two copies of the same gene.
But now let's suppose that a mutation in the active site of B
changes its behavior, making it a little less likely to cut fibrinogen
and a little more likely to activate protease A. In essence,
this would change the relationship between these previously duplicate
genes to something like this:
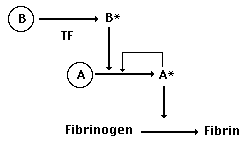
Suddenly, the ability of A to bind to
TF becomes much less important. If B can saturate all of the
available TF-binding sites itself (by virtue of its EGF domain),
then the TF-mediated activation of B, combined with B's affinity
for A, will result in a rapid activation of A, producing plenty
of activated A to convert fibrinogen into clottable fibrin. Sounds
good. But why would natural selection favor a mutation like this
in B's active site? Simple: it would increase the efficiency
of the clotting process by producing a 2-level cascade. Look
closely, and you'll see that our 1-step clotting system required
a direct interaction with TF to activate each protease. The new
2-step system allows each TF to activate a protease B, each of
which in turn can activate scores or hundreds of A's. With so
many more active proteases in the neighborhood of the injury,
clotting can now occur more quickly, increasing the chances of
surviving a hemorrage. Exactly the sort of stuff that natural
selection favors.
Step back for a second and think about
what we've just seen. A simple gene duplication sets the stage
for the selection of active site mutations that would dramatically
improve the clotting process. Gene duplications are neutral mutations,
the sort that occur all the time and therefore, given enough
time, are highly probable. Once the duplication has taken place,
any mutation in the active site that shifts the preferences of
the active site in the direction I have mentioned will be strongly
favored. And that means that a true 2-step system will evolve
very quickly.
Two additional points have to be mentioned.
The first one is obvious. If gene duplication and subsequent
mutation of the duplicate protease can change a 1-step system
into a 2-step one, they could certainly change a 2-step system
into a 3-step one. This means that increases in biochemical complexity
are not only accomodated by evolutionary theory, they are actually
predicted by it. The second point is a little more subtle. Early
stages in the evolution of a clot forming-system are bound not
to work very well. But as the system starts to work better, as
it increases in complexity and efficiency, it begins to present
a danger to the organism. That danger, simply put, is that clotting
might get out of hand. As the clot-forming cascade evolves larger
and larger, there is a chance that a small stimulus will start
a reaction that might cause all of an organism's blood
to clot, or at least enough of it to cause serious problems.
Does evolution have an answer to that, too?
Well, it turns out that it does. First,
keep in mind that a primitive clotting system, adequate for an
animal with low blood pressure and minimal blood flow, doesn't
have the clotting capacity to present this kind of a threat.
But just as soon as the occasional clot becomes large enough
to present health risks, natural selection would favor the evolution
of systems to keep clot formation in check. And where would these
systems come from? From pre-existing proteins, of course, duplicated
and modified. The tissues of the body produce a protein known
as a1-antitrypsin which binds to the active site of serine
proteases found in tissues and keeps them in check. So, just
as soon as clotting systems became strong enough, gene duplication
would have presented natural selection with a working protease
inhibitor that could then evolve into antithrombin, a
similar inhibitor that today blocks the action of the primary
fibrinogen-cleaving protease, thrombin.
In similar fashion, plasminogen,
the precursor to a powerful clot-dissolving protein now found
in plasma, would have been generated from duplicates of existing
protease genes, just as soon as it became advantageous to develop
clot-dissolving capability.
In short, the key to understanding the
evolution of blood clotting is to appreciate that the current
system did not evolve all at once. Like all biochemical systems,
it evolved from genes and proteins that originally served different
purposes. The powerful opportunistic pressures of natural selection
progressively recruited one gene duplication after another, gradually
fashioning a system in which high efficiences of controlled blood
clotting made the modern vertebrate circulatory system possible.
Mining the Biochemical Past
Can we know for sure that this is how
blood clotting (or any other biochemical system) evolved? The
strict answer, of course, is we cannot. The best we can hope
from our vertebrate ancestors are fossils that preserve bits
and pieces of their form and structure, and it might seem that
their biochemistry would be lost forever. But that's not quite
true. Today's organisms are the descendents of that biological
(and biochemical) past, and they provide a perfect opportunity
to test these ideas.
Even a general scheme, like the one I've
just presented, leads to a number of very specific predictions,
each of which can be tested. First, the scheme itself is based
on the use of well-known biochemical clues. For example, most
of the enzymes involved in clotting are serine proteases, protein-cutting
enzymes so-named because of the presence of a highly reactive
serine in their active sites, the business ends of the protein.
Now, what organ produces lots of serine proteases? The
pancreas, of course, which releases serine proteases to help
digest food. The pancreas, as it turns out, shares a common embryonic
origin with another organ: the liver. And, not surprisingly,
all of the clotting proteases are made in the liver. So, to "get"
a masked protease into the serum all we'd need is a gene duplication
that is turned on in the pancreas' "sister" organ.
Simple, reasonable, and supported by the evidence.
Next, if the clotting cascade really
evolved the way I have suggested, the the clotting enzymes would
have to be near-duplicates of a pancreatic enzyme and of each
other. As it turns out, they are. Not only is thrombin homologous
to trypsin, a pancreatic serine protease, but the 5 clotting
proteases (prothrombin and Factors X, IX, XI, and VII) share
extensive homology as well. This is consistent, of course, with
the notion that they were formed by gene duplication, just as
suggested. But there is more to it than that. We could take one
organism, humans for example, and construct a branching "tree"
based on the relative degrees of similarity and difference between
each of the five clotting proteases. Now, if the gene duplications
that produced the clotting cascade occurred long ago in an ancestral
vertebrate, we should be able to take any other vertebrate and
construct a similar tree in which the relationships between the
five clotting proteases match the relationships between the human
proteases. This is a powerful test for our little scheme because
it requires that sequences still undiscovered should match a
particular pattern. And, as anyone knows who has followed the
work in Doolittle's lab over the years, it is also a test that
evolution passes in one organism after another.
There are many other tests and predictions
that can be imposed on the scheme as well, but one of the boldest
was made by Doolittle himself more than a decade ago. If the
modern fibrinogen gene really was recruited from a duplicated
ancestral gene, one that had nothing to do with blood clotting,
then we ought to be able to find a fibrinogen-like gene in an
animal that does not possess the vertebrate clotting pathway.
In other words, we ought to be able to find a non-clotting fibrinogen
protein in an invertebrate. That's a mighty bold prediction,
because if it could not be found, it would cast Doolittle's whole
evolutionary scheme into doubt.
Not to worry. In 1990, Xun Yu and Doolittle
won their own bet, finding a fibrinogen-like sequence in the
sea cucumber, an echinoderm. The vertebrate fibrinogen gene,
just like genes for the other proteins of the clotting sequence,
was formed by the duplication and modification of pre-existing
genes.
Now, it would not be fair, just because
we have presented a realistic evolutionary scheme, supported
by gene sequences from modern organisms, to suggest that we now
know exactly how the clotting system has evolved. That
would be making far too much of our limited ability to reconstruct
the details of the past. But nonetheless, there is little doubt
that we do know enough to develop a plausible and scientifically
valid scenario for how it might have evolved. And that scenario
makes specific predictions that can be tested and verified against
the evidence.
References:
Doolittle, R. F., (1993) "The evolution
of vertebrate blood coagulation: A case of yin and yang,"
Thrombosis and Haemostasis 70: 24-28.
Doolittle, R. F., and Feng, D. F., (1987)
"Reconstructing the evolution of vertebrate blood coagulation
from a consideration of the amino acid sequences of clotting
proteins," Cold Spring Harbor Symposia on Quantitative Biology
52: 869-874.
Doolittle, R. F., and Riley, M. (1990)
"The amino-acid sequence of lobster fibriongen reveals common
ancestry with vitellogenin." Biochemical and Biophysical
Research Communications. 167: 16-19.
Xu, X., and Doolittle, R. F., (1990)
"Presence of a vertebrate fibrinogen-like sequence in an
echinoderm." Proceedings of the National Academy of Sciences
(USA) 87: 2097-2101.
|
